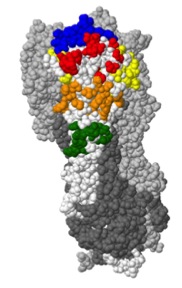
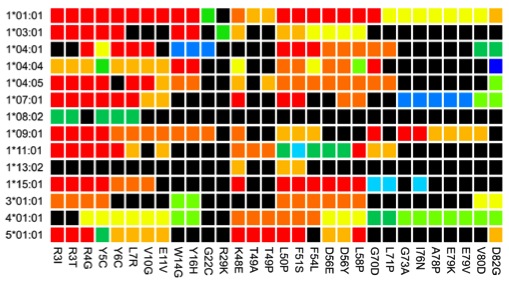
Overview
Computational immunology (or immunoinformatics) is the application of computational methods to research problems in the field of immunology. The Shepherd Group uses diverse computational methods — ranging from molecular dynamics to deep learning — to elucidate (mainly) human immune responses to pathogens, protein therapeutics and cancer.
Key research themes are:
- The analysis of adaptive immune repertoires — large sets of B-cell receptor (antibody) and T-cell receptor sequences serived from Next Generation Sequencing. We host and contribute to the ongoing development of OGRDB: the Open Germline Receptor Database (Lees, et al., NAR, 2020) and VDJbase: the adaptive immune receptor Genotype and Haplotype database (Omer et al., NAR, 2020). Currently we are developing novel deep learning methods to address key challenges, such as how to identify the subset of receptors targeted at a specific antigen (e.g. virus). We are also active members of The AIRR Community (part of The Antibody Society), developing tools and resources to support the analysis of B-cell and T-cell receptor repertoire data.
- Using molecular dynamics (MD) simulations to elucidate the function of important molecules. For example, we've use MD shed light on whether mutations in the stalk of haemagglutinin may enable influenza A virus to escape from broadly-neutralising antibodies (Lees et al., Frontiers in Immunology, 2017) and to gain insight into the dynamic properties of the hepatitis C virus cell entry machinery (Stejskal et al., PLOS Computational Biology, 2020).
- Exploring the boundary between self and non-self. For example, we have developed a novel method called proteome scanning for predicting whether small sequence differences between an individual's endogenous Factor VIII and therapeutic Factor VIII are likely to break self-tolerance (Hart et al., Haematologica, 2019).
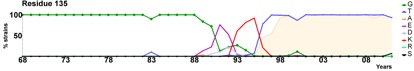
Some images from past research
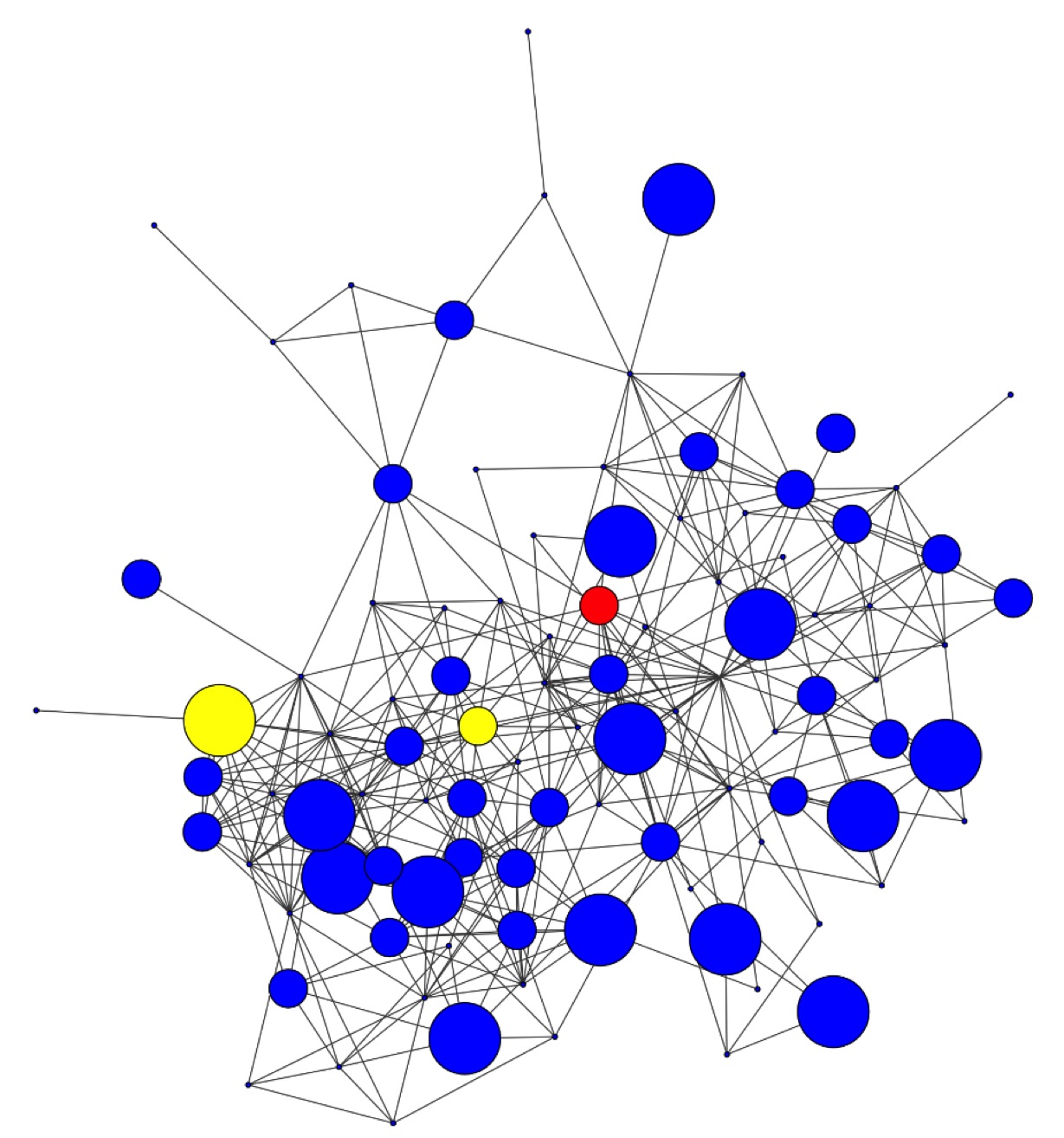
Analysis of an
antibody repertoire after
vaccination. Left: Network
diagram showing the abundance of a small subset of heavy chain
CDR3s in the vicinity of a neutralising antibody of
interest. Sequences identified using the AbMining ToolBox
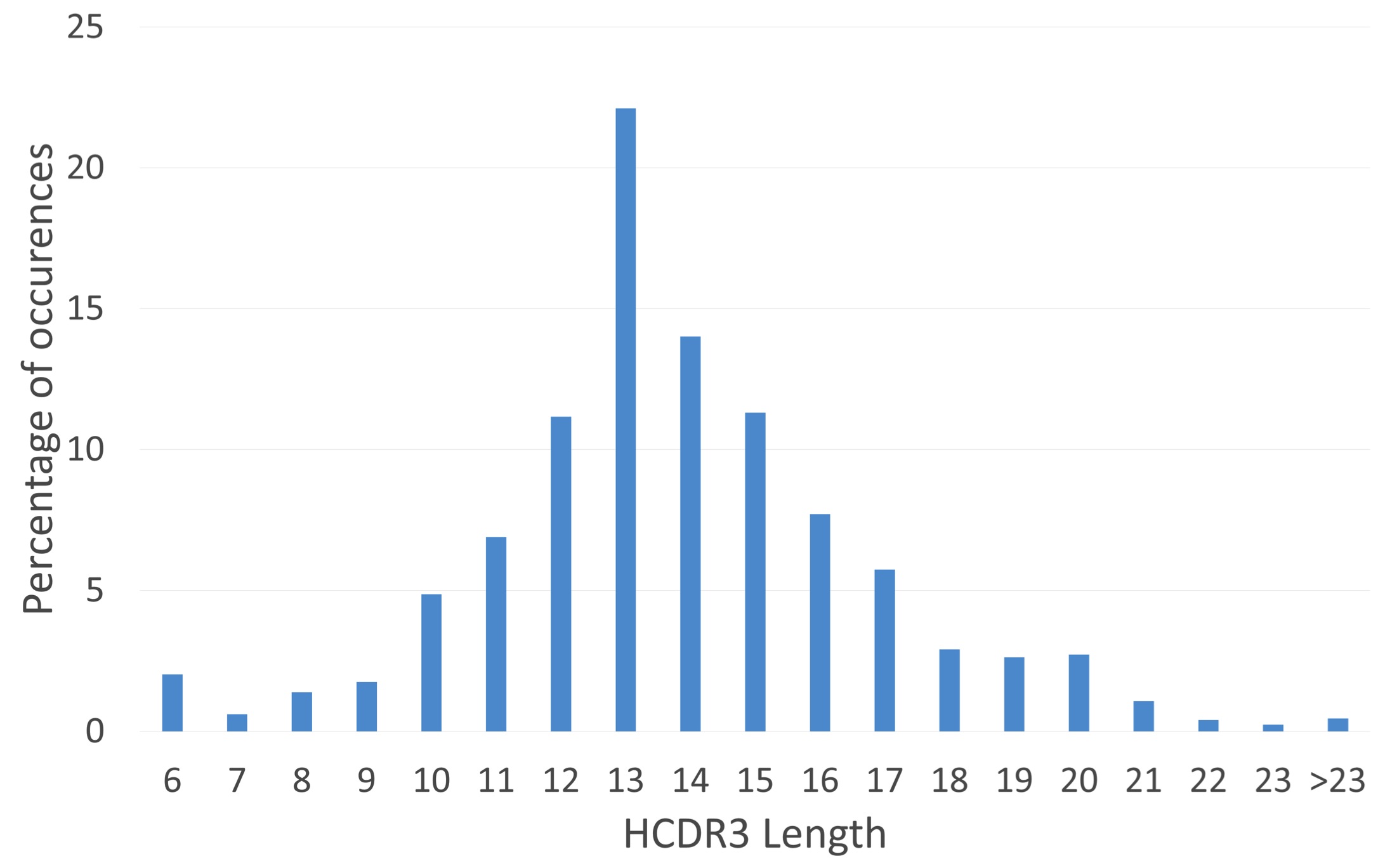
(D'Angelo et al., 2014). Centre: Length
distribution of heavy chain CDR3s. Right: Lineage
plot showing light chain CDR3s after sequence assembly.